Analyzing NASA Spaceflight Microgravity Effects on Mouse Antibody Repertoire
Try HeavyIQ Conversational Analytics on 400 million tweets
Download HEAVY.AI Free, a full-featured version available for use at no cost.
GET FREE LICENSE[Special thanks to Jacci Cenci, Senior Solutions Architect at NVIDIA, Dr. Trisha Rettig and Bailey Bye from Kansas State University for the edits on this blog post.]
Over the last 40 years, NASA has completed over 135 spaceflights. That’s more than 198,700 person-hours, which roughly translates to more than 8,280 days of space travel. More than 830 crew members have taken the ride; some have done so multiple times. During their travels, crew members are exposed to 10 times more radiation than on Earth, as well as microbial pathogens such as Salmonella typhimurium, Pseudomonas aeruginosa, and Candida albicans. In the absence of gravity, improper tailward fluid shifts are also observed in crew members, which can cause swollen heads and ineffective healing, especially in the lower body. Scientists at NASA have been working hard to analyze the causes in order to minimize the negative health impact of deep space missions on astronauts’ health.
Research conducted on the immune responses of mice shows that microgravity causes detrimental effects to the immune system, along with behavioral changes and altered immunized responses. In addition to fluid shift, some possibilities for this change include stress, radiation, and changes in nutritional intake. One important part of the adaptive immune system impacted is B lymphocytes or “B cells”. B cells produce antibodies, which bind to foreign substances in the body. One particularly important region of the antibody is Complementarity-Determining Region 3 (CDR3), which is important for foreign substance binding and is used to measure antibody repertoire diversity. One question scientists are investigating is whether exposure to microgravity alters CDR3 subpopulations. Right now scientists are facing an uphill battle in tracking the CDR3 changes from the origins because they are manually assessing observation from spreadsheets and other data sources.
I partnered with Jacci Cenci, Sr. Solutions Architect, NVIDIA, to introduce scientists at NASA and Kansas State University (KSU) to GOAi (the GPU Open Analytics Initiative) which enables end-to-end analytics, including exploration, extraction, preprocessing, model training, and prediction, by keeping the data in a GPU buffer for maximum efficiency. Our hope was that this framework could help Dr. Stephen Chapes, Dr. Trisha Rettig, Bailey Bye, Claire Ward, and Savannah Hlavacek characterize mice repertoire from space flights in a fast and repeatable workflow. In the end, scientists were amazed and delighted to see the fast response times. MapD is a founding and active member of GOAi along with Anaconda, Apache Arrow, H2O.ai, and Graphistry, and we are proud we could help the scientific community.
Science takes time, and data has to be carefully analyzed to see variety and variability of results over time. Even simple characterizations, such as identifying important C-xx-W motifs, take at least a couple of hours, not only because of the vast amount of data but also because it takes time to utilize multiple tools within the currently established workflow. With the help of MapD, which uses the unique parallel processing power of GPUs, we can see the same insights within a framexwork that returns results in a matter of seconds.
Dataset Description and Objectives
The dataset used in the project was obtained from current research on mice subjected to a physiological model of spaceflight, which is unpublished but will eventually be available at NASA Genelab Data Repository. This dataset consists of 8 treatment groups with 4 mice in each group. For each mouse, the frequency of CDR3 amino acid junctions is captured along with treatment group labels. Each symbol of the treatment group labels denotes the presence or absence of suspension, vaccination with Tetanus toxoid, and the use of the adjuvant (immune stimulator) CPG. For example, mice in treatment group “+-+” were suspended via their tails, not immunized with Tetanus Toxoid, and were injected with CPG.
Each B cell’s unique antibody is generated through a process called V(D)J Recombination in which Variable (V), Diversity (D), and Joining (J) gene segments are cut and spliced together to form the antibody structure. The CDR3 amino acid sequence, which consists of part of the V-, the entirety of the D-, and part of the J-gene segments, provides much of the antibody binding specificity.
The top two experimental goals for this dataset were:
- To distinguish the untreated mice from the ones who are treated, within those 8 treatment groups
- Using machine learning, find the clusters that have been treated the same way within the clustered nodes (treatment groups)
We utilized the GOAi platform to perform analysis and data extraction from MapD, preprocess it in Pygdf/Pandas, analyze nodes in Graphistry, train the model to make clusters with H2O’s KMeans, and store the results back in MapD Core. This notebook illustrates the code along with the steps mentioned for the dataset in this post, driven by Docker so that you don’t have to install everything from scratch.
Getting Started
Setup MapD Community Edition (which includes both the MapD Core SQL engine and the Immerse front-end visualization system) and then install pygdf, pymapd, pygraphistry, and h204gpu. MapD GPU accelerated container can also be downloaded from NVIDIA GPU Cloud.
conda install -c conda-forge pymapd
conda install -c gpuopenanalytics/label/dev pygdf
pip install graphistry
pip install h2o4gpu-0.2.0-cp36-cp36m-linux_x86_64.whl
Loading Data
The first step is to import the libraries and load data into MapD using the pymapd pandas dataframe as an input variable. Pymapd’s load_table automatically chooses pyarrow or binary columnar format to insert values into the table.


Initial Analysis
After loading the data in MapD, we use MapD Immerse, which by default starts on https://localhost:9092, to analyze the dataset. The capability to display charts from different tables in one dashboard, which I’ve shown here, is limited to MapD Immerse Enterprise edition, but you can use the Community Edition to create separate dashboards for each source.

We see 4,128,122 records with 3 feature columns (amino acid junction, frequency, and mouse ID), and a class variable sequence. The distribution of AA junction frequency across the dataset is Right-Skewed and Unimodal; the mean (0.00733) is greater than the median (0.00227) which makes the tail extend to the right with few positive outliers. Mouse AOS 70 has the highest number of CDR3s: 19644, followed by AOS 77 with 18049. And mouse AOS 3 has the least number of CDR3s: 6282. Mouse AOS 15 has the maximum frequency of junctions across the dataset, with 0.02.
Extract Data
Using pymapd, data is extracted to the pygdf dataframe using a SELECT statement. We also extracted MapD’s native rowid which contains a virtual id for each row generated. Through rowid we will associate the predicted results with the original data, which is especially helpful when there is no unique identifier for the dataset.

Preprocess Data
The next step is to remove any duplicate instances of AA junctions. We also need to make sure there are not any null values in the dataset. Then we will capture the position of each amino acid’s position in each junction in the CDR3 region, in order to analyze the repertoire changes across treatment groups. A quick reference to single letter codes of each amino acid can be found here. The helper function below accomplishes this task, and loads data back in MapD:



With the location of each amino acid we can distinguish mice from each other. We will use MapD’s cross-filtering functionality to read the behaviors. For example, A (Alanine) has only one instance at the beginning of an AA junction in CDR3, so we can drill down to the mouse and sequence with this unique junction.
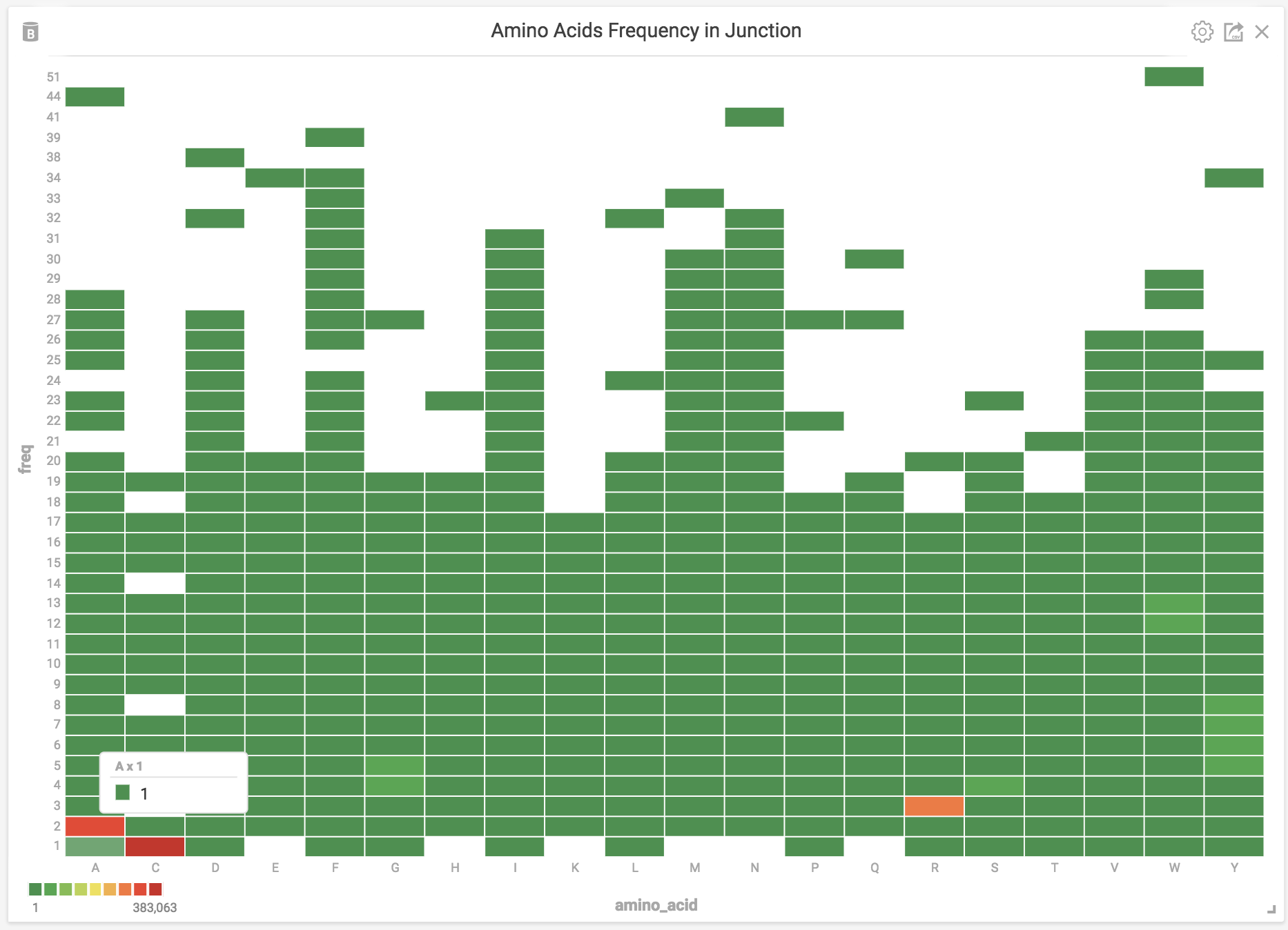

We can see that Mouse 74 exhibits a unique repertoire from the other mice (42, 34, and 18) in the treatment group labeled “---” (control group i.e., no suspension, no tetanus shots, and no CPG). We will use graphistry to further investigate the 72 nodes for this group. Just by looking at the top-level data, we can see the different junctions shared by mice. By drilling down into a treatment group or multiple treatment groups we can begin to analyze their different repertoires.


Now we will label encode categorical columns and split data into 80:20 (train|test) for predictive analysis.


Predictive Model Analysis
Our team used H2O’s KMeans to train the model on GPUs with frequency and aa_junction as features to divide observations into clusters. Finding clusters requires iterative tuning of hyperparameters in order to reach the optimal based upon each dataset. The objective was to make 8 clusters (treatment groups) from the dataset and then evaluate the efficiency of the model.



Model Metrics
Centroids of 8 clusters (treatment groups) obtained from the model can further be optimized by techniques such as Gap Statistic or Silhouette method depending on what platform you’re using, but let’s just stick to the centroids we already have.

Predictions
Assuming we determined the optimal cluster centroids, we can proceed to make clusters on the test set, and based on requirements we can store the results back in MapD.

Conclusion
We did not see any huge changes across treatment groups and the model may need to be further optimized by capturing more continuous variables. But by accelerating machine learning and deep learning research for NASA and KSU, GOAi provides an open source alternative to reduce significant research clock time and computational cost. Utilizing a GPU accelerated pipeline, scientists can focus on reviewing more data from the research. We believe that combining different datasets together in one analytics platform shifts the focus towards further analysis and deeper insights.
Try It Out
You can download the Docker version of the Jupyter notebook demo here. Let us know what you think, on our community forums, or on GitHub. You can also download a fully featured Community Edition of MapD, which includes the open source MapD Core SQL engine, and our MapD Immerse data exploration UI.